
Section 524B of the Federal Food, Drug, and Cosmetic Act changed the regulatory landscape for medical device cybersecurity in a way that guidance documents never did. Guidance is advisory. 524B is law. As of March 2023, the FDA has the authority to refuse to accept premarket submissions for cyber devices that do not include adequate cybersecurity documentation. Understanding what that means for your pen testing program is not optional.
What 524B Actually Requires
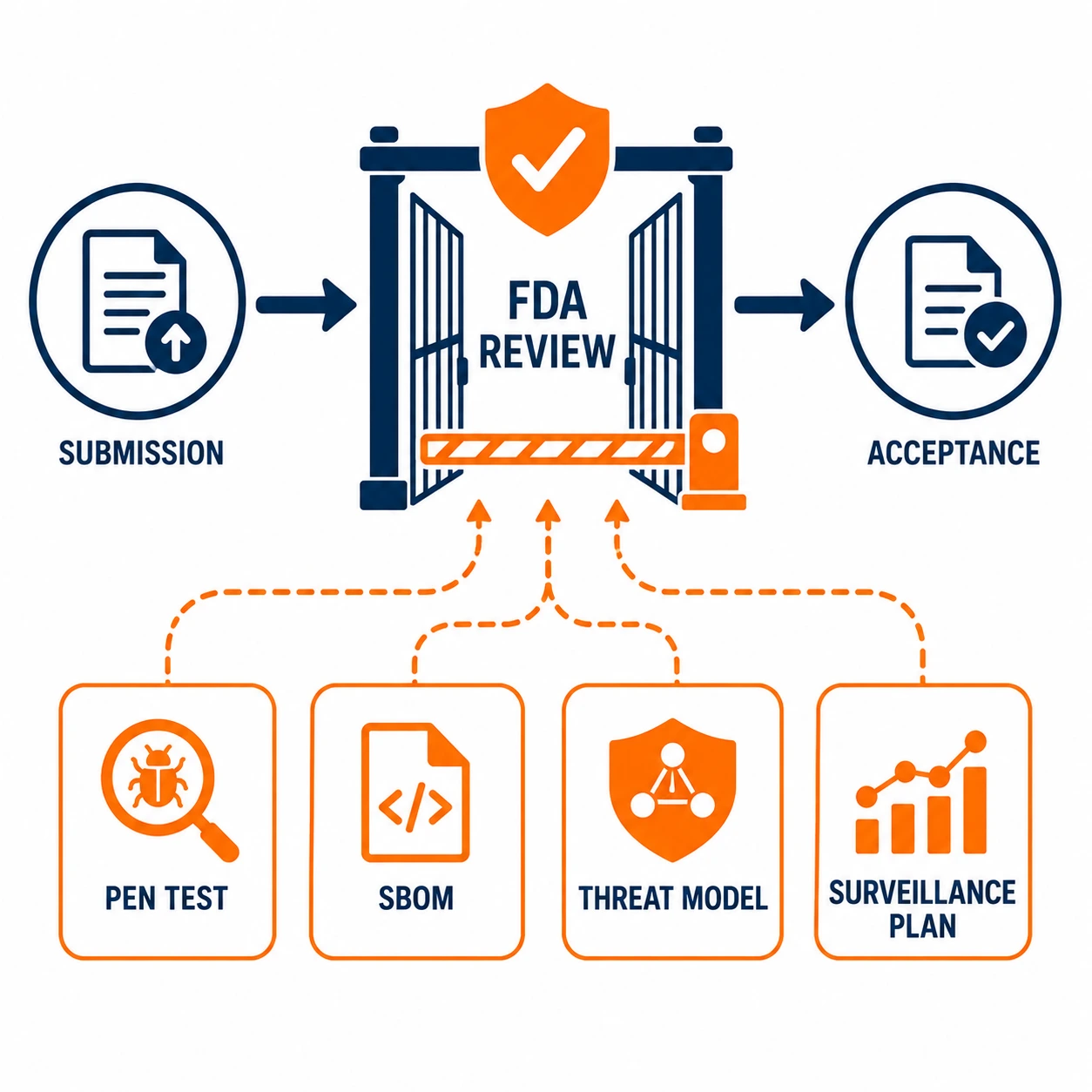
Section 524B requires manufacturers of cyber devices to submit cybersecurity information as part of premarket submissions, establish processes to monitor and address postmarket cybersecurity vulnerabilities, and provide a software bill of materials covering commercial, open-source and off-the-shelf components. Penetration testing is a core component of the premarket cybersecurity evidence the FDA expects to see.
The FDA's Refuse to Accept policy means that submissions without adequate cybersecurity sections are rejected before substantive review begins. That is a significant shift from the prior environment, where cybersecurity deficiencies typically surfaced as questions during review rather than grounds for outright rejection at intake. The bar for what constitutes adequate has been raised, and pen testing is part of meeting it.
Which Devices Are Covered
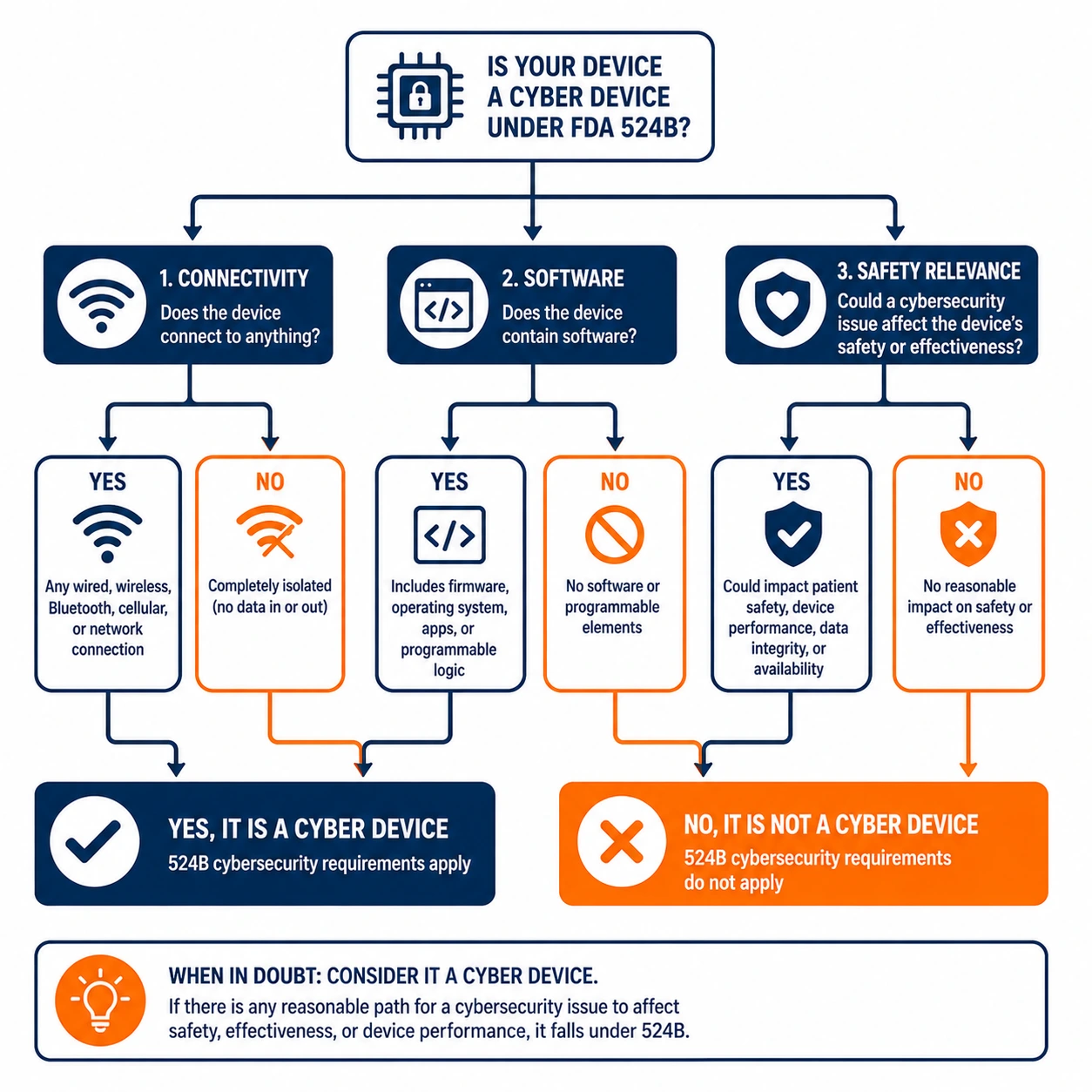
A cyber device under 524B is any device that includes software validated, installed or authorized by the sponsor, has the ability to connect to the internet, and contains any such technological characteristics that could be vulnerable to cybersecurity threats. That definition covers a wide range of devices including infusion pumps, patient monitors, imaging systems and any device with network connectivity or software that affects safety or effectiveness.
Manufacturers should not assume that limited connectivity excludes a device from 524B scope. Bluetooth-only devices, devices that connect only to a proprietary network and devices with indirect internet connectivity through a gateway have all been treated as cyber devices in FDA review contexts. When in doubt, the safer assumption is that 524B applies.
How to Scope a Pen Test for FDA Premarket Submission
The scope of a 524B pen test must reflect the device's intended use environment and its actual attack surface. That means testing hardware interfaces including USB ports, debug interfaces and physical access points. It means testing software and firmware for known vulnerabilities, authentication weaknesses and logic flaws. It means testing communication interfaces including wireless protocols, Bluetooth implementations and any network connectivity the device supports.
The intended use environment matters because it shapes the threat model. A device used in an ICU connected to a hospital network faces a different threat profile than a home-use device connecting to a consumer smartphone app. The pen test methodology should be calibrated to the environment where the device will actually operate, not a generic enterprise IT environment.
What the FDA Looks for in Pen Test Evidence
FDA reviewers evaluating the cybersecurity section of a premarket submission are looking for evidence that the manufacturer understands the device's attack surface, tested against a threat model that reflects real-world adversary behavior, found and addressed vulnerabilities before submission, and has a plan to manage vulnerabilities discovered after clearance.
The pen test report needs to demonstrate all four of these things, not just document what the testers did. That means the report must connect to the threat model, reference the SBOM for context on tested components, document findings with enough detail to show they were genuinely assessed and not just noted, and map to the design controls and risk management file that FDA reviewers will cross-reference.
Postmarket Obligations
524B does not end at clearance. Manufacturers are required to establish processes to monitor for cybersecurity vulnerabilities and to address them in a reasonable time. The FDA expects a patch management plan as part of the premarket submission, and it expects manufacturers to demonstrate that they have the organizational capability to execute on it postmarket.
Building that capability requires more than writing a policy. It requires a tested process for receiving vulnerability disclosures, assessing their relevance to specific device models, developing and validating patches, and communicating with customers and the FDA when significant vulnerabilities are addressed. Pen testing plays a role in validating that process as well as in the initial premarket submission.














