
The rules of engagement need to reflect pharma-specific constraints that a generic pen test provider may not anticipate. No active testing on production validated systems without an approved test environment. No testing during batch manufacturing runs. Change control documentation for any activity that touches a validated system, even in a test environment. These constraints do not limit what the test can find. They ensure that what the test finds is actually actionable.
Before the Test: What Must Be in Place
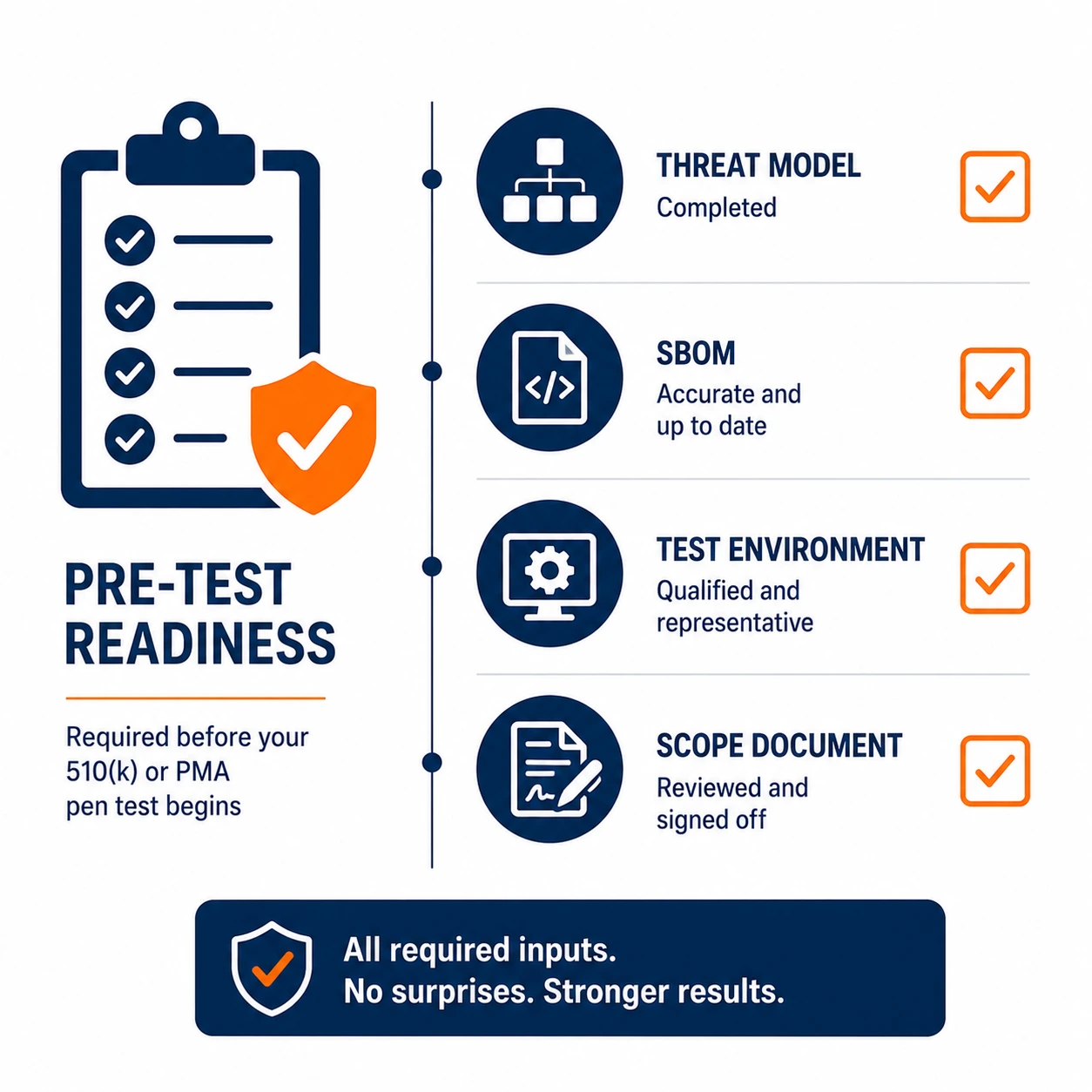
A pen test submitted as FDA cybersecurity evidence is only as credible as the foundation it was built on. FDA reviewers evaluate pen test evidence in the context of the threat model and SBOM that should precede it. A test conducted without a documented threat model looks like a box-checking exercise. A test conducted without a complete SBOM raises questions about whether the tester knew what they were testing.
The threat model must reflect the device's intended use environment and the realistic threat actors relevant to that environment. A patient monitor deployed in an ICU connected to a hospital network faces threats from opportunistic attackers exploiting hospital network access, from insiders with legitimate network presence and from targeted actors interested in patient data or care disruption. The pen test methodology should be calibrated to those threats, not to a generic enterprise IT threat model.
During the Test: What Must Be Covered
FDA guidance identifies specific categories of testing that cybersecurity submissions should address. Hardware interface testing covers physical ports, debug interfaces and any physical access mechanism that could be exploited. A USB port that accepts unsigned firmware updates is a finding. An exposed JTAG interface with no access controls is a finding. These are not theoretical vulnerabilities; they are the kind of hardware-level weaknesses that have appeared in real device compromises.
Software and firmware testing covers known vulnerability validation, authentication mechanisms and update integrity. Can a malicious firmware update be installed without cryptographic verification? Can the device be accessed without valid credentials through an undocumented interface? Does the device's software contain components with known vulnerabilities that have not been patched? Each of these questions needs a documented answer in the submission.
Communication Interface Testing
For most connected devices, communication interface testing is where the most significant findings emerge. Wireless protocol testing covers Bluetooth pairing security, Wi-Fi authentication and any proprietary wireless protocol the device uses. TLS implementation testing validates that encrypted communications are actually secure: certificate validation is enforced, weak cipher suites are not accepted and the device cannot be subjected to downgrade attacks.
The FDA pays particular attention to how devices authenticate to backend systems and how they handle communication with mobile applications. A device that accepts connections from any app presenting the right device identifier without additional authentication creates patient safety risk that reviewers are trained to identify.
After the Test: What the Evidence Package Must Contain
The pen test report submitted to FDA needs to contain more than a standard commercial findings report. It needs to document the scope in terms that map to the device's SSP and threat model. It needs to map each finding to the relevant design control and risk management file entry. It needs to document the remediation status of every finding, including those that remain open with an accepted risk justification.
Unresolved findings are not automatically disqualifying, but they require documented justification. A finding that was assessed, determined to be acceptable given the intended use environment and mitigating controls, and documented as an accepted risk with a compensating control is a defensible position. A finding that appears in the report with no remediation status or justification is not.
The Patch Management Plan
Section 524B requires a postmarket patch management plan as part of the premarket submission. FDA reviewers look for evidence that the manufacturer has a realistic, tested process for monitoring vulnerabilities, assessing their relevance to specific device models and deploying patches in a timeframe proportionate to risk severity. A policy document that describes what the process will be is not the same as evidence that the process works.
Pen testing has a role in validating the patch management process as well as in the initial submission. Testing that a critical vulnerability fix actually closes the vulnerability, rather than just checking a box, is the kind of postmarket validation the FDA expects manufacturers to demonstrate capability for.














